This pipeline gives you a full analysis of nfcore chromatine accessibility peak data (ChIP-Seq, ATAC-Seq or DNAse-Seq) and nfcore RNA-seq count data. It performs DESeq2, TEPIC and DYNAMITE including all preprocessing and postprocessing steps necessary to transform the data. It also gives you plots for deep analysis of the data. The general workflow is sketched in the images below:
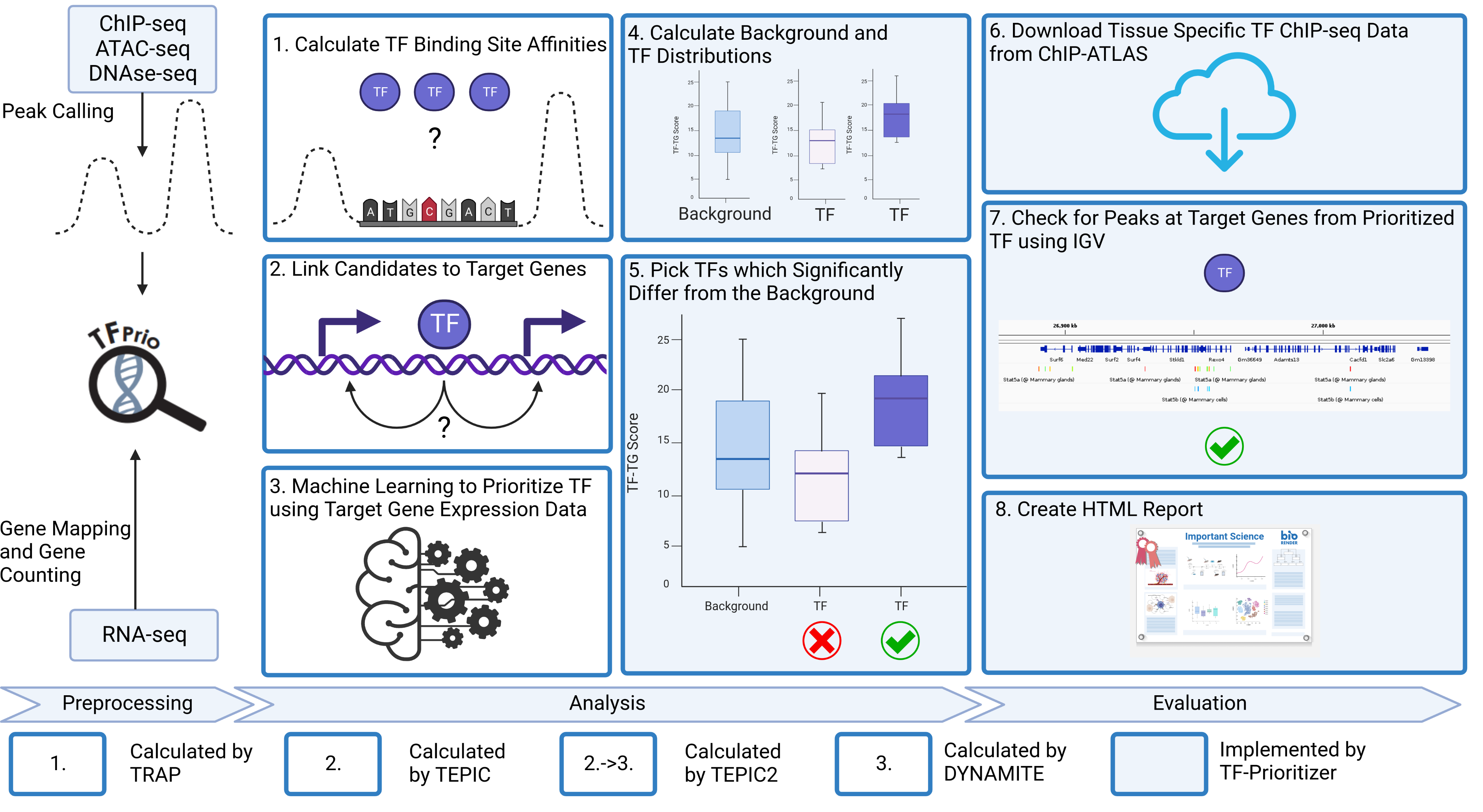

TF-Prioritizer is distributed under the GNU General Public License. The Graphical Abstract and the Technical Workflow was created using biorender.com.
The software can be executed using docker. For the following command, only python3, curl and docker are required. Explanations about the configs can be found in the config readme.
curl -s https://raw.githubusercontent.com/biomedbigdata/TF-Prioritizer/master/docker.py | python3 - -c [config_file] -o [output_dir] -t [threads]Note, that for this approach an internet connection is required. The docker image will be downloaded from DockerHub on the first execution as well as with every update we release. Furthermore, the wrapper script will be fetched from GitHub with every execution.
If curl is not available (for example if you are using windows), or you want to be able to execute the software without an internet connection, you can download the wrapper script from here.
You can then execute the script using
python3 [script_path] -c [config_file] -o [output_dir] -t [threads]We do not recommend using the pipeline without docker, because the dependencies are very complex, and it is very hard to install them correctly. However, if you want to use the pipeline without docker, you can do so by installing the dependencies manually. The dependencies and their correct installation process can be derived from the Dockerfile and the environment scripts which can be found in the environment directory.