

© 2017 Merly Escalona ([email protected]), Sara Rocha, David Posada
University of Vigo, Spain (http://darwin.uvigo.es)
NGSphy is a Python open-source tool for the genome-wide simulation of NGS data (read counts or Illumina reads) obtained from thousands of gene families evolving under a common species tree, with multiple haploid and/or diploid individuals per species, where sequencing coverage (depth) heterogeneity can vary among species, individuals and loci, including off-target and uncaptured loci.
NGSphy simulates reads (or read counts) from alignments originated from single gene trees or gene-tree distributions (originated from species-tree distributions). It is designed to read directly from SimPhy (a simulator of gene family evolution) in the case of gene-tree distributions, but it can also be fed with gene trees directly. These trees can contain orthologs, paralogs and xenologs. Alignments are simulated using INDELible and can represent multiple haploid and/or diploid individuals per species. Then, either Illumina reads (using ART) or read counts are simulated for each individual, with the depth of coverage allowed to vary between species, individuals and loci. This flexibility allows for the simulation of both off-target (captured but not targeted) and uncaptured (targeted but not captured) loci.
You will need a NGSphy settings file and the required files according to the input mode selected (see bellow).
- Examples of setting files can be found here.
- For installation please go here
- For detailed explanations please search in the full manual.
- In the Wiki you can find:
- tutorials for each of the possible input modes
- a validation test
- a use case test
[Single gene-tree scenario]
- NGSPhy settings file
- INDELible control file
- Newick file with single gene tree
- ancestral sequence file (FASTA) (optional)
- reference allele file (optional)
[Species-tree scenario]
- coverage variation
- CSV
- plain text
- log files
- bash scripts
- inputmode 1: allows you to generate DNA sequences from a single gene tree, generate haploid or diploid individuals (by random mating within the same species) and produce reads or read counts [Tutorial 1].

- inputmode 2: allows you to simulate data from a single gene tree and a known ancestral sequence. DNA sequences are evolved from the ancestral sequence under the specified gene-tree, haploid or diploid individuals and reads or read counts generated [Tutorial 2].

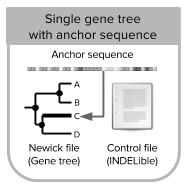
- inputmode 3: allows you to simulate reads/read counts from a single gene tree and a known anchor (tip) sequence. Tree is re-rooted in the anchor sequence before the simulation of DNA sequences [Tutorial 3].
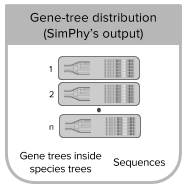
- inputmode 4: this mode uses the output from SimPhy to generate reads or reads counts. SimPhy generates distributions of gene trees and species trees under some desired conditions. Each species tree is here considered a replicate. Given replicates with the same number of tips for all the gene trees, within NGSphy you can then filter the species tree replicates if the contained gene trees do not match your requirements for downstream analyses (eg. number of gene-tree tips per species and your (even) requirement if you want to simulate diploids) [Tutorial 4].
NGSphy can introduce coverage heterogeneity at three different levels: experiment, individual and locus-wide, according to to user-defined statistical distributions, One can also try to mimic the variation in coverage expected for targeted sequencing, including off-target loci, non-captured loci and taxon-specific effects (more details on Manual - Section 6.3). These parameters can be used to recreate different experimental situations:
- NGS run effects: where we expect to obtain different coverages for different runs (replicates), and variation of coverage across individuals and across loci. within single runs.
[Available for all input/simulation modes]
- Targeted sequencing: where some target loci are not captured (non-capture loci), some are captured with different coverage but were not targeted (off-target loci).
[Only possible for input mode 4 (gene tree distributions (SimPhy data))]
- Taxon-specific effects: they can be used, for example, to emulate a decay in coverage related to the phylogenetic distance of the species of interest to the reference sequence used to build the target/capture probes.
[Available for all input/simulation modes]
NGSphy does not have a Graphical User Interface (GUI) and works on the Linux/Mac command line in a non-interactive fashion.
usage: ngsphy [-s <settings_file_path>]
[-l <log_level>] [-v] [-h]
-
Optional arguments:
-s <settings_file_path>, --settings <settings_file_path>:- Path to the settings file. This is optional, by default NGSphy looks for a
settings.txtfile in the current working directory. You can also specify a particular settings file with:
ngsphy -s my_settings.txt- Path to the settings file. This is optional, by default NGSphy looks for a
-l <log_level>, --log <log_level>: Specified hierarchical log levels that will be shown through the standard output. A detailed log will be stored in a separate file. Possible values:DEBUG: shows very detailed information of the program's process.INFO(default): shows only information about the state of the program.WARNING: shows only system warnings.ERROR: shows only execution errors.
-
Information arguments:
-v, --version: Show program's version number and exit.-h, --help: Show help message and exit.
-
If you use NGSphy, please cite:
- Escalona, M, Rocha S and Posada D. (2018) NGSphy: phylogenomic simulation of NGS data. Bioinformatics; doi: https://doi.org/10.1093/bioinformatics/bty146
- Sukumaran, J and Holder MT. (2010). DendroPy: A Python library for phylogenetic computing. Bioinformatics 26: 1569-1571.
-
if running ART cite also:
- Huang W, Li L, Myers JR and Marth, GT. (2012) ART: a next-generation sequencing read simulator. Bioinformatics 28 (4): 593-594
-
if using SimPhy cite also:
- Mallo D, De Oliveira Martins L and Posada D. (2016). SimPhy : Phylogenomic Simulation of Gene, Locus, and Species Trees. Systematic Biology 65(2): 334-344.
-
if using single gene tree inputs, cite also:
- Fletcher, W and Yang Z. (2009) INDELible: A flexible simulator of biological sequence evolution. Molecular Biology and Evolution. 26 (8): 1879–88.