
scGate is an R package that automatizes the typical manual marker-based approach to cell type annotation, to enable accurate and intuitive purification of a cell population of interest from a complex scRNA-seq dataset, without requiring reference gene expression profiles or training data. scGate works with any scRNA-seq technology and with other single-cell modalities.
scGate builds upon UCell for robust single-cell signature scoring and Seurat, a comprehensive and powerful framework for single-cell omics analysis.
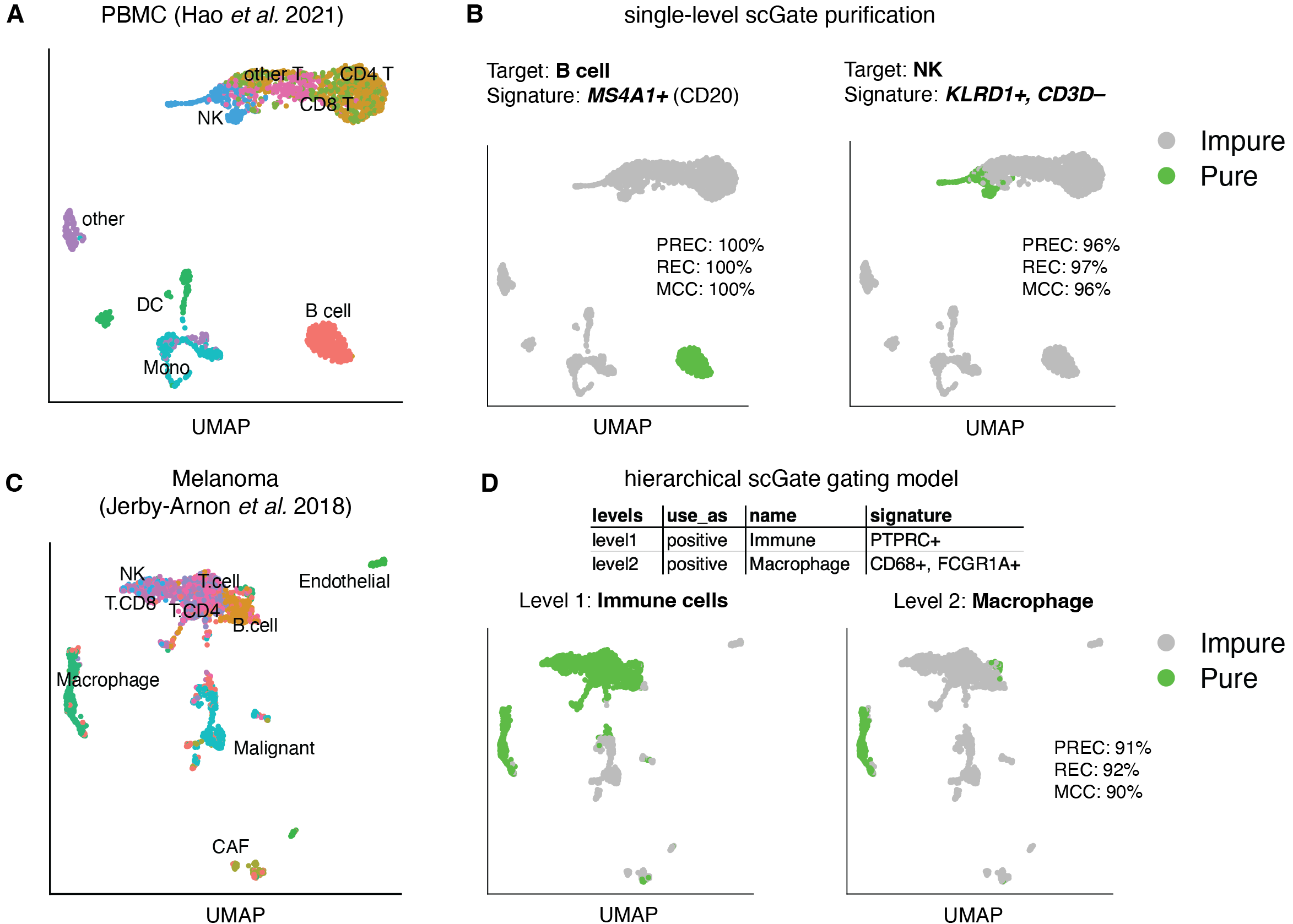
Briefly, scGate takes as input: i) a gene expression matrix stored in a Seurat object and ii) a “gating model” (GM), consisting of a set of marker genes that define the cell population of interest. The GM can be as simple as a single marker gene, or a combination of positive and negative markers. More complex GMs can be constructed in a hierarchical fashion, akin to gating strategies employed in flow cytometry.
scGate evaluates the strength of signature marker expression in each cell using the rank-based method UCell, and then performs k-nearest neighbor (kNN) smoothing by calculating the mean UCell score across neighboring cells. kNN-smoothing aims at compensating for the large degree of sparsity in scRNA-seq data. Finally, a universal threshold over kNN-smoothed signature scores is applied in binary decision trees generated from the user-provided gating model, to annotate cells as either “pure” or “impure”, with respect to the cell population of interest.
Install scGate from CRAN
install.packages("scGate")For older versions of scGate, download a tagged version from GitHub:
library(remotes)
remotes::install_github("carmonalab/scGate", ref="v1.6.2")Use scGate to purify a cell population of interest using manually defined marker genes
library(scGate)
#Get a test scRNA-seq dataset (as a list of Seurat objects)
sample.data.seurat.list <- scGate::get_testing_data()
seurat_object <- sample.data.seurat.list$Satija
#Manually define a simple scGate gating model to purify eg. Natural Killer (NK) cells, using a positive marker KLRD1 and negative marker CD3D
my_scGate_model <- gating_model(name = "NK", signature = c("KLRD1","CD3D-"))
#scGate it!
seurat_object <- scGate(data = seurat_object, model = my_scGate_model)
#Use Seurat to visualize "Pure" and "Impure" cells
DimPlot(seurat_object, group.by = "is.pure")
#Use Seurat to subset pure cells
seurat_object_purified <- subset(seurat_object, subset = `is.pure` == "Pure" )Check out this scGate demo for a reproducible analysis, construction of hierarchical gating models, tools for performance evaluation and other advanced features. More demos for running scGate on different single-cell modalities are available at scGate.demo repository.
A database of gating models for scGate is available on scGate_models and can be loaded using get_scGateDB()
#Get scGate database of pre-defined gating models
scGate_models_DB <- get_scGateDB()
#For example, filter abT cells using one of scGate pre-defined gating models
seurat_object <- scGate(seurat_object, model = scGate_models_DB$human$generic$Tcell.alphabeta)
DimPlot(seurat_object)The first time you run get_scGateDB() the database will be downloaded from the repository. On successive calls it will load your local version of the DB.
You may manually edit the available models (eg in Excel) or create new models for your cell type of interest. You can then load your custom model into R using:
my_scGate_model <- load_scGate_model("path_to_my.model")You can use the plot_tree function to visualize the hierarchical structure of one of the models (requires ggparty).
install.packages("ggparty")
scGate::plot_tree(scGate_models_DB$human$generic$Tcell.alphabeta)scGate can also be used a cell type classifier, to annotate multiple cell types in a dataset. To annotate a dataset with marker-based cell type definitions, simply provide a list of models to scGate, e.g.:
models.list <- scGate_models_DB$human$generic[c("Bcell","MoMacDC","CD8T","CD4T","Erythrocyte")]
obj <- scGate(obj, model = models.list)scGate also provides collections of gating models that work nicely with each other to annotate cell types in specific tissues, e.g. for the tumor micro-environment (TME):
models.TME <- scGate_models_DB$human$TME_HiRes
obj <- scGate(obj, model=models.TME)
table(obj$scGate_multi)See examples of scGate as a classifier at: scGate multi-class and scGate on integrated objects
scGate can be applied to modalities other than RNA-seq, such as ATAC-seq (scATAC-seq demo) and antibody-derived tags (ADT) (CITE-seq demo).
Massimo Andreatta, Ariel J Berenstein, Santiago J Carmona, scGate: marker-based purification of cell types from heterogeneous single-cell RNA-seq datasets, Bioinformatics, Volume 38, Issue 9, 1 May 2022, Pages 2642–2644, https://doi.org/10.1093/bioinformatics/btac141